
MD Simulations of SARS-CoV-2 Spike
SARS-CoV-2 Spike Protein: Molecular Dynamics Simulations of Sequence Variants Compared to the Wild Type Protein
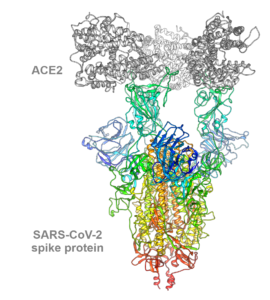 The ongoing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has changed everyday life around the world and, in recent months, more and more virus variants have been discovered. The B.1.1.7 variant of the SARS-CoV-2 virus detected in September 2020 in the United Kingdom shows enhanced infectiousness and thereby replaced the wild type virus variant, leading to increasing patient numbers in affected areas. The trimeric spike glycoprotein of SARS-CoV-2 specifically binds with its receptor-binding domains (RBDs) to the host cell receptor angiotensin-converting enzyme 2 (ACE2) to gain entry into a cell to initiate infection. In the B.1.1.7 variant, mutations within this trimeric SARS-CoV-2 spike protein affect inter-monomeric contact sites within the trimer as well as the receptor-binding domain interacting with the human ACE2-receptor. However, the molecular consequences of mutations within B.1.1.7 on spike protein dynamics and stability or ACE2 binding were largely unknown.
The ongoing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has changed everyday life around the world and, in recent months, more and more virus variants have been discovered. The B.1.1.7 variant of the SARS-CoV-2 virus detected in September 2020 in the United Kingdom shows enhanced infectiousness and thereby replaced the wild type virus variant, leading to increasing patient numbers in affected areas. The trimeric spike glycoprotein of SARS-CoV-2 specifically binds with its receptor-binding domains (RBDs) to the host cell receptor angiotensin-converting enzyme 2 (ACE2) to gain entry into a cell to initiate infection. In the B.1.1.7 variant, mutations within this trimeric SARS-CoV-2 spike protein affect inter-monomeric contact sites within the trimer as well as the receptor-binding domain interacting with the human ACE2-receptor. However, the molecular consequences of mutations within B.1.1.7 on spike protein dynamics and stability or ACE2 binding were largely unknown.
In this research project, we perform all-atom molecular dynamics (MD) simulations in order to compute the 3-dimensional motions in the SARS-CoV-2 wild type spike protein and compare them with the B.1.1.7 variant. At the atomic level, our investigations will reveal regions within the spike protein trimer with an altered structural flexibility, contact rearrangements between the three monomers building the trimeric spike and whether the structural RBD–ACE2 interface is changed and the binding affinity to the human receptor is affected by the mutation in the receptor-binding domain. We anticipate that our findings will be starting points for further characterization of other emerging virus variants.
Publication: Paper
Eileen Socher, Marcus Conrad, Lukas Heger, Friedrich Paulsen, Heinrich Sticht, Friederike Zunke, Philipp Arnold